Command Line Syntax
dnenrich <opt> num_perm alias.list gene_size.matrix gene.sets mutation.list {--weightSet,SET_OF_INTEREST,WEIGHT} {background.list1 background.list2 ...}"<opt>" indicates the required options argument, one of those specified here.
"{}" indicates optional arguments ("--weightSet" recommended for advanced users only).
num_perm: Number of permutations to run, where in each permutation the de novo mutations are randomly re-distributed within their corresponding classes (i.e., base context and functional impact), proportional to the gene sizes for that class.
alias.list: Official gene names with aliases that map back to the gene name. The gene sets and mutation lists must have the same gene names. Aliases come from the HGNC database of human gene names.

Gene alias file format is defined here.
gene_size.matrix: Matrix of gene sizes calculated, for all exonic genes. The first 2 columns contain the group information and the mutation type, and then each subsequent column contains the size of the gene in the corresponding column header.
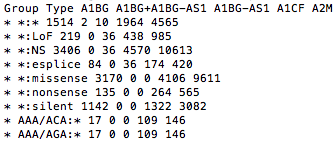
Gene size matrix format is defined here.
gene.sets: Tab delimited file listing all genes in the gene set the user is testing. It is crucial that this file is TAB DELIMITED.
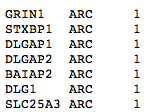
Gene set format is defined here.
mutation.list: File listing the mutations in the users dataset, with 5 columns.

Mutation list format is defined here.
background.list: List of genes to be "conditioned on" during permutation, listed as one gene per row.
Background list format is defined here.
The following table lists the command line options for DNENRICH analysis (to replace "<opt>" on the command line).
The user can specify any combination of options (excluding ".", which means no other options should be given). For example, replace "<opt>" with "xp" to count multiple mutations in the same gene in the same individual, as well as outputting the genes impacted by the simulated mutations.
| Option | Description |
|---|---|
| . | None of the options below are applied. |
| x | Allow gene-level recurrence for the same "Individual" (person). i.e., for when individuals IDs for mutations are unknown (if individuals are "pooled"). |
| p | Print out each set of permuted de novo mutations (genes hit) to standard out. |
| E | Calculate the fraction of null replicates where the test statistic exactly matches the observed test statistic ("# of mutations").
This option is recommended for advanced users only. |
| --weightSet,SET,WEIGHT | Used to up-weight genes in the gene set by a specified factor (and then tests only that set).
Useful in the following case, for example, to assign a weight to a set and assess how many de novos would be expected under the up-weighted effective gene sizes? This option is recommended for advanced users only. |
The output (written to standard output) contains text describing the assessed significance of the mutations having fallen in each of the gene sets given in the input.
NOTE that standard error contains useful log information but should be captured separately from standard out, e.g., as described here.

For each gene set present in the input list, it contains the following information (in rows NOT starting with "__GRP1"):
| Column number | Explanation |
|---|---|
| 1 | Gene set |
| 2 | Number of de novos in gene for it to be included in the assessment ("*" denotes 1 or more, i.e., enrichment; "2" would denote a test of recurrence within that gene set) |
| 3 | P-Value |
| 4 | Observed (weighted) number of mutations |
| 5 | Expected (weighted) number of mutations |
| 6 | Number of total genes in the gene set |
| 7 | Number of mutations in input file |
For each gene set, the rows starting with "__GRP1" give the details of the mutations actually present in the gene set:
| Column number | Explanation |
|---|---|
| 1 | __GRP1 |
| 2 | Gene set |
| 3 | Individual with mutation in gene in the set |
| 4 | User-defined weight of the mutation in this individual |
| 5 | Ordinal index of this mutation among all mutations overlapping some gene (in the gene set) in this individual. This will only be >1 for "individuals" with more than 1 mutation in the same gene, though mutations overlapping multiple genes that are in this same gene set will correspondigly get counted multiple times here (though not for assessment of significance). |
| 6 | Ordinal index of this mutation among all genes (in the gene set) affected by the mutation (for mutations overlapping multiple genes) in this individual. This will only be >1 for "individuals" with single mutations hitting multiple genes in the gene set. |
| 7 | Total number of mutations this individual has (in the input mutation list), irrespective of it being in this gene set or not |
| 8 | Name of gene in gene set in which individual bears mutation |
| 9 | Weight of this gene in the gene set |
Running the extractDnenrichResults.csh script on the output from dnenrich will format the results to yield a more nicely-formatted output file containing the 11 columns described below.
This assumes you've named the output file res-GENE SETS-DISEASE-MUTCLASS, where for example we named the output file "res-kirov-SCZ-NS", where "kirov" is the name of the gene sets being tested for mutations found in the "SCZ" data set of "NS" type mutations (see example here).
| Column number | Column heading | Explanation |
|---|---|---|
| 1 | GROUP | String describing who you are testing, taken from the name of the file |
| 2 | MUT_TYPE | String describing the mutation type, taken from the name of the file |
| 3 | NUM_MUTS | Number of mutations present in the set |
| 4 | SET_CONTEXT | Set context |
| 5 | SET | Name of the gene set you are testing |
| 6 | P_VAL | P-value |
| 7 | NGENES_SET | Number of genes in the gene set |
| 8 | OBS_HITS_STAT | Observed number of mutations (when the weights of the genes are all 1), or actual number of mutations hitting the gene set |
| 9 | EXP_HITS_STAT | Expected number of mutations hitting the gene set |
| 10 | NUM_MUTS_HIT | Actual number of mutations |
| 11 | GENES | Genes in the gene set hit by mutations of this type (and counts of mutations for genes hit more than once) |